

- NAME: Amiram Goldblum
- POSITION TITLE: Professor Emeritus of Medicinal Chemistry, HUJI
Biographical Sketch
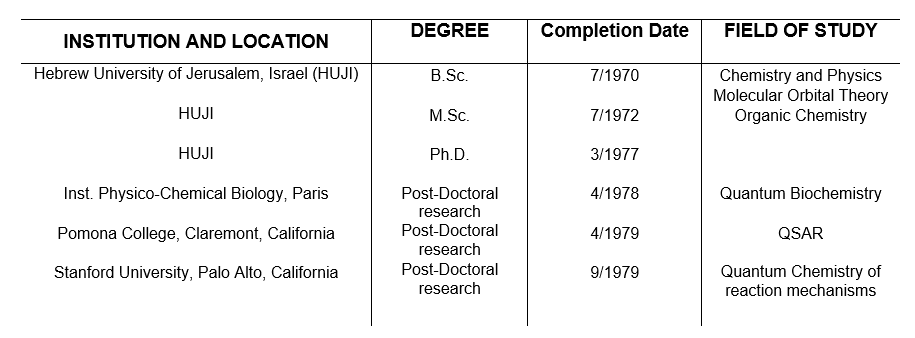
Education/Training
Personal Statement
My studies reflect a diverse background which allowed me to develop research programs and original ideas in the field of molecular discovery and design. I studied Chemistry and Physics for B.Sc. followed by a M.Sc. focused on Molecular Orbital theory (1972 with Prof. A.D. Bergman and Dr. A. Meyer) and a mechanistic organic chemistry thesis (1977, with Prof. Rafael Mechoulam), at the Hebrew University of Jerusalem. Subsequently, I went for a postdoctoral position on Quantum Biochemistry at the Institute for Physico-Chemical Biology, Paris (1977-1978) with Profs. Alberte and Bernard Pullman. That was followed by a second PostDoc on Quantitative Structure-Activity Relations at Pomona College, California (1978-1979) with Prof. Corwin Hansch, and a third PostDoc on Quantum Mechanical computations of reaction pathways at Stanford University with Dr. Gilda Loew (1979). I spent sabbatical leaves as visiting scientist at Stanford Research Institute, Menlo Park California (Summers of 1982, 1983, 1985, 1986, 1987), Roswell Part Memorial Institute, Buffalo NY (1984), National Cancer Institute Frederick MD (1993), Cambridge University UK (Tom Blundell 2006), and Imperial College London (Michael Sternberg 2010 and 2012)
I Served as Chair of the Medicinal Chemistry Department (2006-2009). Currently, I am head of the Molecular Modeling and Drug Design unit at the Institute for Drug Research of the Hebrew University. Developed an awarded algorithm for solving extremely complex combinatorial problems (American Chemical Society National Meeting, COMP division, 2000) that has been applied to many of my group's studies in the last 20 years –for issues of Structural Biology, Protein-ligand and Protein-Protein interactions, Molecular Design of small molecules, of peptides and of Proteins, and more recently, applied to Cheminformatics and informatics. Together with some 15 PhD students and ~25 M.Sc students supervised directly by me, those recent applications already provided unprecedented accuracy in predicting drug candidates, in producing patents as well as in forming the basis for the construction of two start-up drug discovery companies - Pepticom (for the design of peptide drug candidates and RebioticsRX (for developing nanoliposomal drug delivery). Our CB1R antagonists have been recently licensed to BioNanoSim, Jerusalem.

Positions and Honors

Contributions to Science
1. Protein structure, conformations and interactions
The need to devise a novel computational method emerged from our studies of protein structure and conformations that are based on the atomic positions reported in protein crystal structures from X-ray experiments in the Protein Data Bank. As only heavy atom positions are reported, it is required to positions protons in order to have the full atomic positions which is required for computing interactions. Proton positions on rotatable bonds – such as those of Tyrosine, Threonine, Serine and water molecules depend on each other in a combinatorial manner. Therefore positioning of rotatable protons in protein structures from X-rays is an extremely complex combinatorial problem, which we decided to solve by a new idea for solving such problems in a generic solution. Our first version of the new algorithm (Proteins 2000) was used to compare predicted positions to those of neutron diffraction studies. Subsequently, the algorithm was further developed to enable the solution of immense combinatorial problems of size 10100 (PNAS 2002). Further applications for conformational studies of proteins included cyclic peptides of different sizes as well as construction of a kinase homology model with six large loops that were constructed simultaneously including conformational variations. Finally , we adapted our algorithm (now called “Iterative Stochastic Elimination”, ISE) to dock flexible ligands into flexible binding sites in proteins. In all these publications, we used energy based criteria to prioritize and to sort the many options.
• Glick, M and Goldblum, A. A novel energy-based stochastic method for positioning polar protons in protein structures from X-rays. Proteins-Structure Function and Genetics 2000 38, 273-287 https://doi.org/10.1007/978-1-4615-4141-7_117 • Glick, M., Rayan, A., & Goldblum A. A stochastic algorithm for global optimization and for best populations: A test case of side chains in proteins, Proceedings of the National Academy of Sciences of the United States of America, 2002 99(2):703–708. http://doi.org/10.1073/pnas.022418199 • Rayan, A , Senderowitz, H and Goldblum, A. Exploring the conformational space of cyclic peptides by a stochastic search method , J. Mol. Graph. Model. 2004 22(5) ,319-333 http://doi:10.1016/j.jmgm.2003.12.012 • Rayan, A, Noy, E, Chema, D, Levitzki, A and Goldblum, A. Stochastic algorithm for kinase homology model construction Curr. Med. Chem. 2004 11(6) 675-692 http://doi:10.2174/0929867043455701 • Noy, E., Tabakman, T. and Goldblum A. Constructing ensembles of flexible fragments in native proteins by iterative stochastic elimination is relevant to protein–protein interfaces, Proteins, 2007 68:702–711. http://doi.org/10.1002/prot.21437 • Gorelik, B. and Goldblum A. High quality binding modes in docking ligands to proteins. Proteins, 2008 71:1373–1386. http://doi.org/10.1002/prot.21847
2. Properties of Molecules
One of the main questions in drug discovery is the issue of “drug likeness” – is it possible to decide whether a molecule could become a drug based on its two-dimensional or three–dimensional structure? Since the publication of Lipinski’s “rule of five” (ROF) in 1997 it became common to use ROF to decide if a molecule could or could not be used as a drug. However, that is totally wrong, as the ROF relates only to the bioavailability of molecules taken orally, and it was shown that a huge number of non-drugs obey those requirements of oral bioavailability. We have therefore applied ISE to that problem of “drug likeness” property as well as to another issue of property of molecules, that depends upon the Boltzmann weighted contributions of different molecules conformations to a molecular property such as dipole moment.
• Rayan A, Marcus D, Goldblum A: Predicting Oral Druglikeness by Iterative Stochastic Elimination. Journal of Chemical Information and Modeling. 2010;50(3):437-445. http://doi.org/10.1021/ci9004354 • Lavy T, Harries D, Goldblum A. Molecular Properties from Conformational Ensembles: 1. Dipole Moments of Molecules with Multiple internal Rotations. J. Phys. Chem. A 2011 115(23): 5794-5809 DOI: 10.1021/jp108837a • Ursu O, Rayan A, Goldblum A, Oprea TI: Understanding drug-likeness. Wiley Interdisciplinary Reviews-Computational Molecular Science. 2011;1(5):760-781 http://doi: 10.1002/wcms.5 • Anwar Rayan, Mizied Falah, Jamal Raiyn, Beny Da'adoosh, Sleman Kadan, Hilal Zaid, and Amiram Goldblum: Indexing Molecules for their hERG liability, European Journal of Medicinal Chemistry 2013 65, 304-314 http:// doi: 10.1016/j.ejmech.2013.04.059
3. Molecular properties for nanoliposomal delivery of drugs
Nanoliposomal delivery has advantages mainly in diseases that are associated with the EPR effect (enhanced permeability and retention therapeutic effective concentrations of a drug, which also is able to be slowly released from the nanoliposome, has the ability to deliver the drug to the site of disease, where the EPR effect is most prominent, and to eradicate the localized disease. A main question is whether one could predict which molecules could be loaded “remotely” (i.e., prepared nanoliposomes with a weak acidic or basic salt immersed into a solution of a amphipathic drug candidate) in such effective concentrations, and whether the resulting contents would be stable enough in plasma. In order to make predictions, we have used first a “decision tree” and subsequently used our ISE to model the remote loading as well as the stability (slow release) of molecules based on experiments performed by us and form literature. Our models for remote loading were confirmed in more than 90% and our models for stability – in more than 80%. We have also found that hundreds of clinical tested drugs are candidates for being remotely loaded nad stable in nanoliposomes, and about 70 of them are FDA approved drugs. Some of them are already tested experimentally, while nano-mupirocin is a new systemic delivery vehicle for an antibiotic drug that was only used externally (batroban). Here are some of the main publications
• Zucker D, Marcus D, Barenholz Y and Goldblum A. Liposome drugs' loading efficiency: a working model based on loading conditions and drug's physicochemical properties. J. Control. Release 2009 139(1) 73-80 http:// doi: 10.1016/j.jconrel.2009.05.036 • Cern A, Golbraikh, A, Sedykh, A, Tropsha A., Barenholz Y and Goldblum A. Quantitative Structure – Property Relationship Modeling of Remote Liposome Loading Of Drugs, J. Control. Release 2012 160(2) 147-157 http: doi: 10.1016/j.jconrel.2011.11.029 • Cern A, Barenholtz Y, Tropsha A, and Goldblum A: Computer-aided design of liposomal drugs: in silico prediction and experimental validation of drug candidates for liposomal remote loading, Journal Control. Release 2014 173, 125-131 http://doi.org/10.1021/ci9004354 • Cern A, Nativ-Roth E, Goldblum A, Barenholz Y. Effect of solubilizing agents on mupirocin loading into and release from PEGylated nanoliposomes. J Pharm Sci. 2014 Jul;103(7):2131-8. http://doi: 10.1002/jps.24037 • Cern A, Michael-Gayego A, Bavli Y, Korena E, Goldblum A, Moses AE, Xiong YQ, Barenholz Y. Nano-mupirocin: enabling the parenteral activity of mupirocin. Eur. J. Nanomed. 2016 8 (3) 139-149 DOI: 10.1515/ejnm-2016-0006 • Cern A, Marcus D, Tropsha A, Barenholz Y and Goldblum A. New Drug Candidates for Liposomal Delivery Identified by Computer Modeling of Liposomes' Remote loading J. Control. Release 2017 252, 18-27 DOI: 10.1016/j.jconrel.2017.02.015
4. Drug Candidates and properties from ligand based modeling
Most of our recent contributions are the result of collaborations with experimentalists who need molecular solutions for dealing with disease conditions. In some other cases we initiate such modeling efforts due to our own interests and seek for collaborations with experimentalists. All our predictions must be tested and all our publications combine the predictions with the results of testing them in a“wet” lab.
• Froese, DS et al. Structural basis of glycogen branching enzyme deficiency and pharmacologic rescue by rational peptide design, Human Molecular Genetics 2015 24, (20) 5667-567 http://doi: 10.1093/hmg/ddv280 • Basu A, Sohn, YS, Alyan, M , Nechushtai R, Domb AJ and Goldblum, A. Discovering Novel and Diverse Iron-Chelators in Silico, J. Chem. Inf. Model. 2016 56 (12), 2476-2485 DOI: 10.1021/acs.jcim.6b00450 • Zatsepin M, Mattes A, Rupp S, Finkelmeeier D, Basu A, Burger-Kentischer A and Goldblum A. Computational Discovery and Experimental Confirmation of TLR9 Receptor Antagonist Leads, J. Chem. Inf. Model. 2016, 56(9) 1835-1846 DOI: 10.1021/acs.jcim.6b00070 • Da’adoosh B, Marcus D, Rayan A, King F, Che JW and Goldblum A. Discovering highly selective and diverse PPAR-delta agonists by ligand based machine learning and structural modeling, Scientific Reports 2019 9, Art. Number 1106 DOI: 10.1038/s41598-019-38508-8 • El-Atawneh S, Hirsch S, Hadar R, Tam J and Goldblum A. Prediction and Experimental Confirmation of Novel Peripheral Cannabinoid-1 Receptor Antagonists. 2019, 59, 3996-4006 DOI: 10.1021/acs.jcim.9b00577

Research Support and/or Scholastic Performance
Current support
